Antibiotics and Antivirals
Penicillin went from laboratory curiosity to mass production in under a decade. It saved millions of lives in World War II and transformed medicine. Within three years of its clinical introduction, Alexander Fleming was already warning: use it carelessly, and bacteria will evolve around it. He was right. The question now is whether we can slow the clock.
Practise this lesson
Four printable worksheets that build from the foundations up to exam-style questions — start at whatever level suits you.
In 1945, Alexander Fleming accepted the Nobel Prize and warned: "There is the danger that the ignorant man may easily under-dose himself and by exposing his microbes to non-lethal quantities of the drug make them resistant."
Before reading: at the evolutionary level, why does exposing bacteria to a non-lethal dose of an antibiotic make them resistant? What process is occurring, and why does incomplete treatment accelerate it?
Know
- How antibiotics work — mechanisms of action
- Why antibiotics cannot treat viral infections
- How antivirals work and their limitations
- The mechanisms by which bacteria develop antibiotic resistance
Understand
- Why antibiotic resistance is an evolutionary process, not a personal one
- Why incomplete antibiotic courses accelerate resistance development
- Why developing new antivirals is harder than developing antibiotics
Can Do
- Explain the evolution of antibiotic resistance using natural selection
- Distinguish antibiotic and antiviral mechanisms of action
- Evaluate strategies for managing antibiotic resistance
Core Content
Selective toxicity: hit what bacteria have and we don't
Antibiotics are compounds that kill or inhibit the growth of bacteria. They achieve this by targeting structures or processes that are essential to bacteria but absent or different in human cells — this selective toxicity is what makes them safe for human use.
Cell wall synthesis inhibition
Protein synthesis inhibition
DNA replication/transcription inhibition
Cell membrane disruption
Metabolic pathway inhibition
What to write in your book
- Antibiotics work by selective toxicity — they hit structures bacteria have but human cells don't.
- Five target classes: cell wall (penicillins), 70S ribosome (tetracyclines/macrolides), DNA gyrase (fluoroquinolones), cell membrane (polymyxins), folate synthesis (sulfonamides).
- Bactericidal = kills bacteria directly; bacteriostatic = halts growth so the immune system clears them.
Penicillins and vancomycin work by inhibiting synthesis of _____, the polymer that forms the bacterial cell wall.
Annotated Diagram — Antibiotic Targets in a Bacterial Cell
Pattern A — Draw and Annotate
In your book, draw a labelled diagram of a bacterial cell and annotate it to show where five different classes of antibiotic act. Your diagram must include:
- The cell wall — label with the antibiotic class that targets it and explain the mechanism.
- The cell membrane — label with the antibiotic class and explain how disruption causes cell death.
- The ribosome (70S) — label with two different antibiotic classes targeting the 30S and 50S subunits respectively.
- The DNA/chromosome — label with the antibiotic class targeting DNA replication.
- A metabolic pathway box — label with the antibiotic class targeting folate synthesis.
- A note explaining why each target is safe for human cells (i.e. what is different about human cells that makes each drug selective).

Antibiotics exploit differences between bacterial and human cells — targeting cell walls, ribosomes, DNA replication and metabolic pathways that bacteria have but we do not.
No bacterial structures = no antibiotic targets
This is one of the most clinically important — and most frequently misunderstood — points in infectious disease. Antibiotics are completely ineffective against viral infections. The reason is straightforward: viruses do not have the structures that antibiotics target.
- Viruses have no cell walls (no peptidoglycan to inhibit)
- Viruses do not have their own ribosomes — they hijack the host cell's 80S ribosomes to make proteins (antibiotic ribosomal targets do not apply)
- Viruses do not independently replicate their DNA — they use host cell enzymes (bacterial gyrase targets do not apply)
- Viruses do not synthesise folate or have independent metabolic pathways to target
When a patient with a viral infection (influenza, common cold, COVID-19, most sore throats) receives antibiotics, the antibiotic does nothing to the virus. It does, however, kill susceptible bacteria in the patient's gut microbiome — reducing the diversity of their normal flora and creating opportunities for resistant bacteria to establish. This is a significant driver of antibiotic resistance in the community.
What to write in your book
- Antibiotics don't work on viruses — viruses have no cell wall, no 70S ribosomes, no DNA gyrase, no folate pathway.
- Viruses use the host cell's own machinery, so antibiotic targets simply don't exist.
- Taking antibiotics for a viral infection gives no benefit but still drives resistance by killing gut flora.
Antibiotics cannot treat viral infections because:
Antibiotic Resistance by Natural Selection
Targeting the few processes unique to the virus
Developing antiviral drugs is fundamentally harder than developing antibiotics. Bacteria are cells — distinct from human cells, with their own structures to target. Viruses use the host cell's own machinery, making it difficult to hit the virus without also hitting the host.
The strategy for antivirals is to target the few processes that are unique to the virus — typically specific viral enzymes or viral surface proteins.
| Mechanism | Target | Example Drug | Virus Treated |
|---|---|---|---|
| Nucleoside/nucleotide analogues | Viral polymerase — the enzyme that copies viral DNA or RNA. The drug mimics a nucleoside, gets incorporated into the growing viral genome, and terminates replication | Aciclovir, remdesivir, tenofovir | Herpes (aciclovir); COVID-19 (remdesivir); HIV/Hep B (tenofovir) |
| Neuraminidase inhibitors | Neuraminidase — a surface enzyme influenza uses to release new viral particles from infected cells. Inhibiting it traps new virions on the cell surface | Oseltamivir (Tamiflu), zanamivir | Influenza A and B |
| Protease inhibitors | Viral protease — an enzyme that cleaves viral polyprotein into functional components. Without it, new virus particles cannot mature | Ritonavir, lopinavir; nirmatrelvir (Paxlovid) | HIV; COVID-19 |
| Entry/fusion inhibitors | Viral surface proteins (e.g. gp41 in HIV) or host cell receptors required for viral entry | Enfuvirtide; maraviroc | HIV |
| Reverse transcriptase inhibitors | Reverse transcriptase — the enzyme HIV uses to convert its RNA genome into DNA. Unique to retroviruses — not present in human cells | Zidovudine (AZT), efavirenz | HIV |
What to write in your book
- Antivirals are harder to design than antibiotics because viruses use host machinery — few unique targets exist.
- Key mechanisms: polymerase inhibitors (aciclovir), neuraminidase inhibitors (oseltamivir), protease inhibitors (Paxlovid), reverse transcriptase inhibitors (AZT).
- HIV uses 3+ drug combinations (HAART) so a single virus can't acquire resistance to all targets at once.
Antivirals destroy existing virus particles directly, in the same way antibiotics kill bacteria.
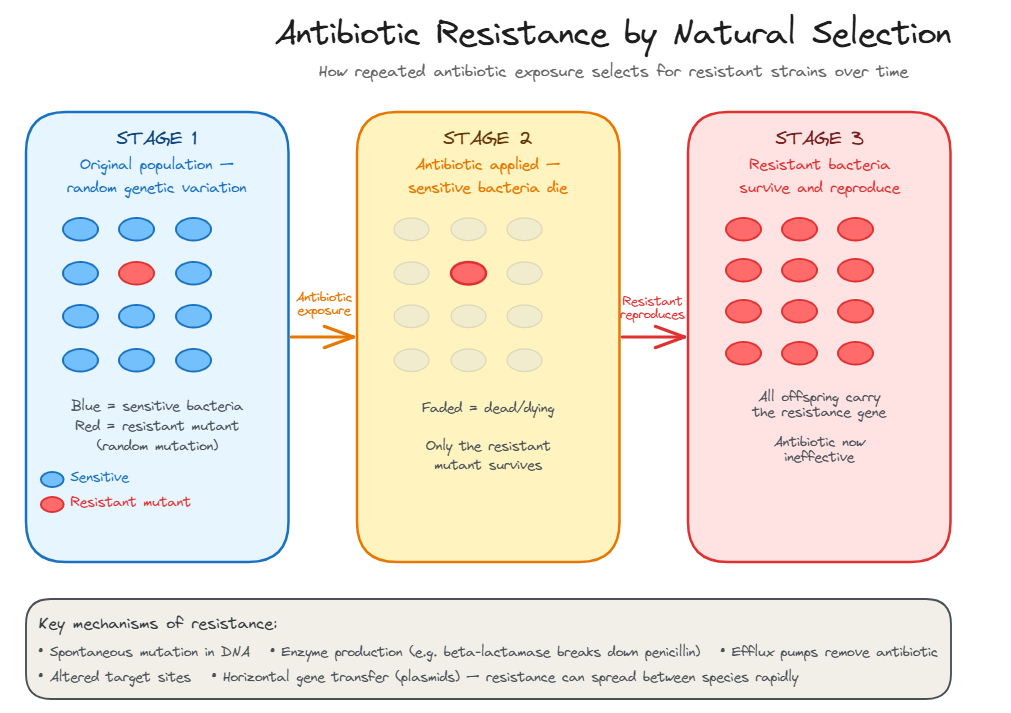
Antibiotic resistance evolves through natural selection when susceptible bacteria are killed and resistant variants survive and reproduce.
Antibiotics are effective against viral infections such as influenza and the common cold.
The antibiotic doesn't create resistance — it selects for it
Antibiotic resistance is one of the clearest examples of natural selection observable within a human lifetime. It is not something that happens to individual bacteria — it is a population-level evolutionary process.
Antibiotic resistance evolves through natural selection — the antibiotic does not create resistance, it selects for pre-existing resistant variants
Mechanisms of Resistance
Bacteria develop resistance through several molecular mechanisms, which can arise through spontaneous mutation or be acquired by horizontal gene transfer (plasmids carrying resistance genes passed between bacteria, even of different species).
What to write in your book
- Resistance is natural selection: rare pre-existing resistant variants survive the antibiotic and reproduce — the population becomes resistant.
- Mechanisms: drug-destroying enzymes (beta-lactamase), altered binding sites (MRSA's PBP2a), efflux pumps, reduced porins, bypass pathways.
- Horizontal gene transfer via plasmids spreads resistance between species in hours — far faster than mutation alone.
A population of bacteria becomes antibiotic-resistant because:
Structured Data Analysis — Antibiotic Resistance Over Time
Pattern A — Structured Data Analysis
The table shows the percentage of Staphylococcus aureus isolates resistant to key antibiotics in Australian hospitals over time.
| Year | Penicillin resistance (%) | Methicillin/oxacillin resistance (%) | Vancomycin resistance (%) |
|---|---|---|---|
| 1945 | 0 | N/A (not yet introduced) | 0 |
| 1950 | 40 | 0 | 0 |
| 1960 | 80 | 2 | 0 |
| 1975 | 90 | 15 | 0 |
| 1990 | 95 | 30 | 0 |
| 2005 | 96 | 42 | 0.1 |
| 2020 | 97 | 28 | 0.3 |
- Describe the trend in penicillin resistance from 1945 to 2020. At what point did the rate of increase slow, and suggest why.
- Methicillin was introduced in 1960 as a penicillin-resistant replacement. Using the data and your knowledge of natural selection, explain the rise in methicillin resistance between 1960 and 1990.
- Vancomycin resistance remained at 0% until after 2000, despite vancomycin being used since the 1950s. Suggest two reasons why vancomycin resistance took so much longer to emerge than penicillin or methicillin resistance.
- Methicillin resistance declined slightly from 42% (2005) to 28% (2020). Propose a biological and a public health explanation for this decline.
- Using this data, evaluate the claim: "We will eventually run out of effective antibiotics for treating S. aureus infections." In your evaluation, refer to the mechanisms of resistance and the current drug development pipeline.
MRSA (methicillin-resistant Staphylococcus aureus) emerged in the 1960s — just one year after methicillin was introduced as a penicillin-resistant replacement. It is now endemic in hospitals globally and is a leading cause of hospital-acquired infection. MRSA requires treatment with vancomycin — one of the last-line antibiotics. Vancomycin-resistant strains (VRSA) have since appeared.
You will analyse antibiotic resistance data and evaluate management strategies in Activity 2 and Short Answer Q3.
How Antibiotics Work
- Target structures present in bacteria but not human cells.
- Cell wall (peptidoglycan) — penicillins, vancomycin.
- Ribosomes (70S bacterial vs 80S human) — tetracyclines, macrolides.
- DNA gyrase — fluoroquinolones.
- Folate synthesis — sulfonamides (bacteria synthesise folate; humans do not).
Why Antibiotics Don't Work on Viruses
- Viruses have no cell walls, no ribosomes, no folate synthesis.
- They use host cell machinery — the antibiotic's targets don't exist.
- Using antibiotics for viral infections promotes resistance without benefit.
Antibiotic Resistance — Natural Selection
- Resistance pre-exists as rare random mutations in the population.
- Antibiotic acts as selection pressure — kills susceptible, spares resistant.
- Resistant variants reproduce → resistant population dominates.
- Horizontal gene transfer (plasmids) spreads resistance between species.
Managing Resistance
- Complete antibiotic courses — eliminate population, not just reduce.
- Avoid unnecessary antibiotic prescribing (especially for viral infections).
- Use narrow-spectrum antibiotics where possible.
- Antibiotic stewardship programs in hospitals.
- Develop new antibiotics and alternative treatments (phage therapy).
Why Antibiotics Don't Work on Viruses
A fresh set drawn from this lesson's question bank — feedback shown immediately. +5 XP per correct · +25 XP all correct
Pick your answer, then rate your confidence — that tells the system what to drill next.
UnderstandBand 4(3 marks) 1. Explain why antibiotics are effective against bacterial infections but not viral infections. In your answer, refer to at least two specific antibiotic targets and explain why viruses do not possess these targets.
1 mark: general principle — antibiotics target structures unique to bacteria / absent in human cells (and viruses) · 1 mark: target 1 with explanation of why viruses lack it · 1 mark: target 2 with explanation of why viruses lack it
ApplyBand 4(3 marks) 2. Using the concept of natural selection, explain how a population of bacteria can develop antibiotic resistance following exposure to an antibiotic. Refer to the role of random mutation, selection pressure, and reproduction in your answer.
1 mark: random mutation pre-exists in the population before antibiotic exposure · 1 mark: antibiotic acts as selection pressure — kills susceptible, resistant variants survive · 1 mark: resistant survivors reproduce — resistance gene inherited by offspring, resistant population dominates
EvaluateBand 5(4 marks) 3. Evaluate the impact of antibiotic resistance on the treatment of infectious disease. In your answer, refer to the global scale of the problem, the factors that drive resistance, and strategies that can be used to manage it.
1 mark: scale — AMR kills ~1.27 million/year globally; more than HIV or malaria · 1 mark: drivers — overprescribing (especially for viral infections), incomplete courses, agriculture use, horizontal gene transfer · 1 mark: management strategies — antibiotic stewardship, completing courses, narrow-spectrum use, new drug development · 1 mark: evaluative conclusion — serious and worsening threat; manageable but requires sustained global coordinated action
Show all answers
Multiple choice
Q1 — C: Human cells are eukaryotic — they have no cell wall at all. Penicillin targets peptidoglycan synthesis — a structure unique to bacterial cell walls. With no cell wall to inhibit, penicillin has no target in human cells. (B) is wrong — human cells have no cell wall, not a different one. (A) and (D) are biologically incorrect.
Q2 — B: Viruses use host cell machinery — they have no cell wall, no independent ribosomes, and no DNA gyrase. Antibiotics cannot affect viral replication because their targets do not exist in viral particles or in the intracellular environment where viruses replicate. (A) is irrelevant to the mechanism. (C) is incorrect — antibiotics do not destroy the immune system, though they can alter the microbiome. (D) is a misconception — "resistance" in the virological sense does not apply here; the issue is the complete absence of antibiotic targets.
Q3 — D: Resistance evolves through natural selection — pre-existing resistant variants survive the antibiotic and reproduce. Bacteria do not deliberately mutate in response to exposure (A) — mutations are random and pre-exist. Antibiotics do not chemically modify DNA (B). Humans do not become immune to antibiotics (C).
Q4 — A: Neuraminidase cleaves sialic acid residues that tether newly assembled influenza virions to the host cell surface. Without neuraminidase, newly formed virus particles cannot escape the infected cell and are trapped — preventing spread to new cells. (B) describes RNA polymerase inhibitors (e.g. baloxavir). (C) describes haemagglutinin function — which neuraminidase inhibitors do not target. (D) describes protease inhibitors.
Q5 — C: HIV mutates rapidly, and resistance to a single drug can emerge during treatment. Triple therapy dramatically reduces the probability of resistance because a single viral particle would need to simultaneously acquire resistance mutations for three different drugs — a statistically very unlikely event. (B) is partially correct (each drug does target a different step) but does not give the primary reason for combination therapy, which is resistance prevention. (A) is a secondary consideration, not the primary rationale. (D) is partially true for some drugs but not the main reason.
Short Answer Model Answers
SA1: Antibiotics are effective against bacterial infections because they target structures or processes that are essential to bacteria but absent in (or structurally different from) human cells — this selective toxicity allows the drug to kill bacteria without harming the patient. Viruses lack these targets because they are not cells; they use the host cell's own machinery for most functions, leaving very few virus-specific targets for drugs to act on. Target 1 — cell wall synthesis: antibiotics such as penicillins and vancomycin inhibit the synthesis of peptidoglycan — the structural polymer of the bacterial cell wall. Viruses have no cell wall and contain no peptidoglycan. A viral particle entering a cell does not need to maintain a cell wall, so this target simply does not exist in any stage of the viral life cycle. Target 2 — bacterial ribosomes: antibiotics such as tetracyclines (30S subunit) and macrolides (50S subunit) inhibit the bacterial 70S ribosome. Viruses do not have their own ribosomes — they commandeer the host cell's 80S ribosomes to translate viral proteins. Antibiotic ribosomal inhibitors bind specifically to the 70S bacterial ribosome structure; they do not bind effectively to the 80S human ribosome, and since there are no viral ribosomes to target, these drugs have no effect on viral protein synthesis.
SA2: Before any antibiotic is introduced, random mutations during bacterial replication occasionally produce variants with characteristics that confer resistance to that antibiotic — for example, mutations that alter the antibiotic's target site or produce enzymes that inactivate the drug. These resistant variants arise spontaneously and are rare in the bacterial population, but they exist before the antibiotic is ever applied. When the antibiotic is introduced, it acts as a selection pressure: susceptible bacteria — those without the resistance mutation — cannot survive at therapeutic antibiotic concentrations and are killed or prevented from reproducing. Resistant variants are not affected by the antibiotic and continue to survive and reproduce normally. Over successive generations, the antibiotic-susceptible bacteria are progressively eliminated from the population while the resistant variants multiply. Because resistance genes are heritable — passed to daughter cells during binary fission — the offspring of resistant variants also carry the resistance gene. The result is that the bacterial population is increasingly dominated by resistant individuals. The antibiotic has not created the resistance: it has selected for pre-existing variants that happened to carry a useful trait. This is natural selection operating within a bacterial population — the same fundamental process that drives all evolutionary change.
SA3: Antibiotic resistance is one of the most serious global public health threats of the 21st century. Antimicrobial resistance (AMR) was estimated to have directly caused approximately 1.27 million deaths globally in 2019 — exceeding deaths from HIV/AIDS (860,000) or malaria (640,000) in the same year. Without effective antibiotics, routine surgeries (appendectomies, caesarean sections, joint replacements), cancer chemotherapy, and organ transplantation — all of which rely on antibiotics to prevent and treat infections — would become significantly more dangerous. The drivers of resistance are multiple and interconnected. Overprescribing of antibiotics — particularly for viral respiratory infections where they have no effect — exposes bacteria in the patient's microbiome to unnecessary selection pressure. Incomplete antibiotic courses allow the most resistant bacteria in a treated population to survive and repopulate. Agricultural use of antibiotics as growth promoters in livestock exposes large bacterial populations to sub-therapeutic concentrations — one of the most significant drivers of resistance globally. Horizontal gene transfer allows resistance genes to spread between bacterial species far faster than mutation alone would permit. Management strategies include antibiotic stewardship programs in hospitals and primary care — guidelines that reduce inappropriate prescribing, reserve certain antibiotics as last-line treatments, and promote narrow-spectrum over broad-spectrum agents where possible. Patient education about completing prescribed courses and not sharing antibiotics addresses the incomplete course and self-medication problems. Investment in new antibiotic development and alternative treatments — such as bacteriophage therapy, antimicrobial peptides, and monoclonal antibodies against bacterial targets — is critical, though the commercial pipeline remains inadequate. Overall, antibiotic resistance is a serious, worsening, and potentially catastrophic threat that is directly driven by human behaviour — overuse, misuse, and agricultural application. It is manageable if sustained, coordinated global action is taken, but the trajectory of resistance data suggests the window for effective action is narrowing.
Five timed questions on antibiotics, antivirals, and resistance. Beat the boss to bank a tier — gold (perfect + fast), silver (80%+), or bronze (cleared).
⚔ Enter the arenaClimb platforms, hit checkpoints, and answer quick-recall questions on this lesson. Lighter than the boss — pure recall practice.
You were asked why a non-lethal dose of antibiotic makes bacteria resistant — and why incomplete treatment accelerates it.
The mechanism: a non-lethal dose kills the most susceptible bacteria but leaves the less susceptible — those with partial resistance — alive. These survivors experience selection pressure without being eliminated. They reproduce, and their offspring inherit whatever partial resistance they possessed. Repeat the process and the population drifts progressively toward higher resistance. Fleming understood this intuitively in 1945, a decade before we understood the molecular mechanisms.
Incomplete treatment is the same process amplified. When you stop taking antibiotics because you feel better, the bacterial population has been reduced but not eliminated. The surviving bacteria are, by definition, the ones that were hardest to kill — the most resistant members of the population. You then allow them to multiply back to full population size, now with a much higher proportion of resistance.
If you predicted "natural selection" — correct. If you predicted "the bacteria learn to resist" — that is a common misconception. Bacteria do not learn, adapt deliberately, or respond to threat. The resistance was already there in a few individuals before the antibiotic arrived. The antibiotic just made resistance the winning trait.