Biology • Year 12 • Module 8 • Lesson 7
Genetic Diseases: CF, PKU, Huntington's Disease, Type 1 Diabetes
Recall the gene, altered protein, inheritance pattern, and physiological consequences for each of the four genetic diseases studied in this lesson.
1. Label the gene–protein–disease pathway for cystic fibrosis
The image below shows the pathway from the CFTR gene mutation to the physiological consequence of thick mucus in the lungs. Write the missing labels into boxes A–H. Each answer should come from Card 2 of the lesson. 8 marks
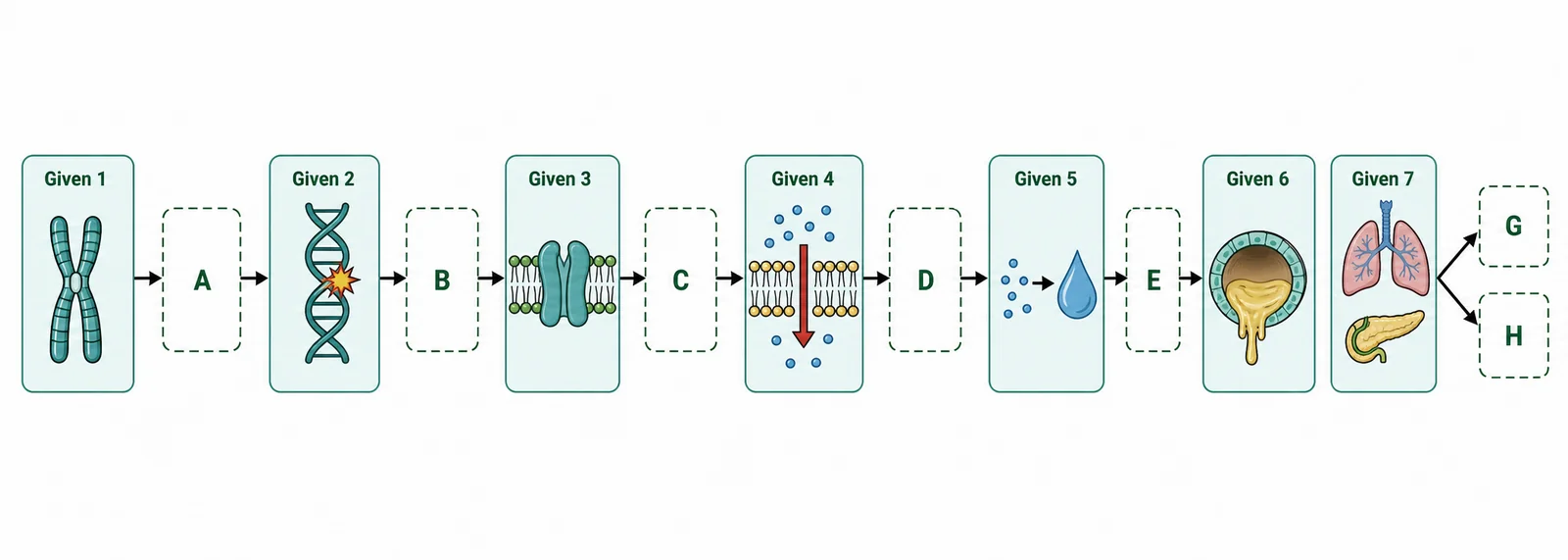
- A, gene name and chromosome: _______________________
- B, most common mutation: _______________________
- C, what happens to the CFTR protein after this mutation: _______________________
- D, normal function of CFTR protein: _______________________
- E, immediate cellular consequence (ion transport): _______________________
- F, osmotic consequence in the lumen: _______________________
- G, physical property of mucus that changes: _______________________
- H, two organ systems most severely affected: _______________________
2. Term–definition match
The ten definitions below are shuffled. Write the matching term from this list in the right-hand column: genetic disease, mutation, autosomal recessive, autosomal dominant, penetrance, phenylalanine hydroxylase, polyglutamine tract, CAG trinucleotide repeat, HLA genes, gain-of-function mutation. 10 marks
| # | Definition (shuffled) | Matching term |
|---|---|---|
| 2.1 | A disease caused by inherited mutations or chromosomal abnormalities; non-infectious and present from conception. | |
| 2.2 | A permanent change in the DNA sequence that may alter protein structure and function. | |
| 2.3 | An inheritance pattern in which two copies of a mutated allele are required for the disease to manifest; carriers are unaffected. | |
| 2.4 | An inheritance pattern in which one copy of a mutated allele is sufficient to cause disease; an affected parent passes a 50% risk to each child. | |
| 2.5 | The proportion of individuals with a disease-causing genotype who actually express the phenotype. | |
| 2.6 | The liver enzyme that converts phenylalanine to tyrosine; non-functional in PKU. | |
| 2.7 | An abnormally long stretch of repeated glutamine amino acids produced by the expanded HTT mutation; this structure is toxic to neurons. | |
| 2.8 | The type of repetitive DNA expansion in the HTT gene that causes Huntington’s disease; repeat count above 36 is disease-causing. | |
| 2.9 | Immune-recognition genes on chromosome 6; specific variants (DR3, DR4) confer predisposition to Type 1 diabetes. | |
| 2.10 | A mutation in which the altered protein acquires a new toxic action rather than simply losing its normal role; explains why Huntington’s disease is autosomal dominant. |
3. True or false, with correction
For each statement, circle T or F. If the statement is false, write the corrected version on the line. 10 marks (1 T/F + 1 correction where needed)
3.1 Cystic fibrosis is caused by the accumulation of thick mucus in the airways. T / F
3.2 A carrier of the CF allele typically displays no symptoms because one functional CFTR allele produces enough chloride channel protein to maintain normal mucus hydration. T / F
3.3 PKU is an autosomal dominant condition in which one mutated PAH allele is sufficient to cause intellectual disability. T / F
3.4 Huntington’s disease typically causes symptoms from birth because the HTT gene mutation is present at conception. T / F
3.5 The approximately 50% concordance for Type 1 diabetes in identical twins shows that genetic predisposition alone is not sufficient to cause the disease, environmental factors are also required. T / F
4. Function recall, what does it do?
Answer each in 1–2 sentences using precise terms from the lesson. 8 marks (2 each)
4.1 What is the normal function of the CFTR protein in epithelial cells, and how does its loss produce thick mucus?
4.2 What is the normal function of phenylalanine hydroxylase (PAH), and why does its absence cause neurological damage in an untreated child with PKU?
4.3 What is the role of pancreatic beta cells in blood glucose regulation, and how does their destruction in Type 1 diabetes produce hyperglycaemia?
4.4 Why is the Australian Guthrie heel-prick test performed within 48–72 hours of birth, and why is early diagnosis of PKU so critical?
5. Fill-the-blank, Huntington’s disease
Complete the paragraph by writing one word or short phrase in each blank. Use the word bank below. 8 marks, 1 per blank
Word bank: gain-of-function • polyglutamine • 36 • autosomal dominant • 50% • striatal • anticipation • CAG
Huntington’s disease is caused by an expansion of ______________ trinucleotide repeats in the HTT gene on chromosome 4. Normal individuals have fewer than ______________ repeats; disease-causing alleles carry 36 or more, producing a mutant huntingtin protein with an abnormally long ______________ tract at its N-terminus. This makes the protein toxic rather than simply non-functional, so Huntington’s is classified as a ______________ mutation. Because one copy of this toxic allele is sufficient to cause the disease, the inheritance pattern is ______________. Each child of an affected parent has a ______________ chance of inheriting the mutation. The disease preferentially destroys ______________ neurons in the basal ganglia, producing the characteristic involuntary movements known as chorea. Because the CAG repeat can expand further during transmission, offspring may show earlier and more severe onset, a phenomenon called ______________.
6. Build a concept map, gene–protein–disease
Draw labelled arrows between the five term chips below to show how they connect for any one of the four genetic diseases studied in this lesson. Each arrow must carry a linking phrase (e.g. “encodes”, “produces”, “causes”). Aim for at least 5 labelled arrows. 5 marks
Supplied terms: mutation · gene · altered protein · physiological consequence · genetic disease.
Q1, CF gene–protein–disease labels
A: CFTR (Cystic Fibrosis Transmembrane conductance Regulator), chromosome 7. B: F508del (deletion of phenylalanine codon at position 508). C: The CFTR protein misfolds and is degraded before reaching the cell membrane, it never functions as a channel. D: CFTR normally acts as a Cl− ion channel in the apical membrane of epithelial cells, secreting chloride ions into the lumen so water follows by osmosis. E: Cl− cannot be secreted into the lumen (the channel is absent from the membrane). F: Without Cl− secretion, water does not follow by osmosis, the lumen becomes dehydrated. G: Mucus dehydrates and becomes thick, viscous, and sticky. H: Lungs (chronic bacterial infections, progressive inflammation) and pancreas (blocked ducts, impaired enzyme secretion, malabsorption).
Q2, Term–definition matches
2.1 genetic disease • 2.2 mutation • 2.3 autosomal recessive • 2.4 autosomal dominant • 2.5 penetrance • 2.6 phenylalanine hydroxylase • 2.7 polyglutamine tract • 2.8 CAG trinucleotide repeat • 2.9 HLA genes • 2.10 gain-of-function mutation.
Q3, True / false with correction
3.1 False. Correction: CF is caused by a mutation in the CFTR gene that prevents the CFTR protein from functioning as a Cl− channel. Thick mucus is the consequence, not the cause. Always start with the gene and protein.
3.2 True.
3.3 False. Correction: PKU is autosomal recessive. Both PAH alleles must be mutated for the disease to manifest. A carrier with one mutated allele still produces enough phenylalanine hydroxylase to metabolise phenylalanine normally and shows no symptoms.
3.4 False. Correction: Huntington’s disease typically does not cause symptoms until 30–50 years of age. The mutation is present from conception, but the toxic mutant huntingtin protein accumulates slowly over decades and neurons die progressively; symptoms only appear after sufficient neuronal loss has occurred.
3.5 True.
Q4.1, CFTR function and thick mucus
CFTR normally acts as a Cl− ion channel in the apical membrane of epithelial cells, secreting chloride ions into the airway lumen; water follows by osmosis, keeping the mucus layer hydrated and able to trap and clear pathogens. When CFTR is absent (as in CF), Cl− is not secreted, water does not follow, and mucus dehydrates, it becomes thick and sticky, obstructing airways and providing a medium for chronic bacterial colonisation.
Q4.2, PAH function and PKU neurological damage
Phenylalanine hydroxylase (PAH) catalyses the conversion of phenylalanine (an essential amino acid from food) to tyrosine in the liver. When PAH is non-functional, phenylalanine cannot be converted and accumulates in blood and tissues to toxic levels. High phenylalanine competes with other amino acids for transport across the blood–brain barrier and disrupts myelin synthesis, killing or impairing developing neurons, producing intellectual disability and seizures if left untreated.
Q4.3, Beta cell function and Type 1 diabetes hyperglycaemia
Pancreatic beta cells in the islets of Langerhans produce and secrete insulin in response to rising blood glucose. Insulin enables cells (especially muscle, liver, adipose) to take up glucose for energy or storage, returning blood glucose to the set point. In Type 1 diabetes, autoimmune destruction of beta cells means no insulin is produced; glucose cannot enter cells and accumulates in the blood, producing chronic hyperglycaemia and eventually life-threatening diabetic ketoacidosis.
Q4.4, Guthrie test and early PKU diagnosis
The Guthrie heel-prick test measures blood phenylalanine concentration from a dried blood spot collected 48–72 hours after birth, enough time for dietary phenylalanine from feeding to accumulate if PKU is present. Early diagnosis is critical because brain damage from phenylalanine accumulation begins within days of birth and is irreversible once it occurs. A child diagnosed at birth and immediately placed on a low-phenylalanine diet develops normally; delayed diagnosis results in permanent intellectual disability.
Q5, Huntington’s cloze
In order: CAG • 36 • polyglutamine • gain-of-function • autosomal dominant • 50% • striatal • anticipation.
Q6, Sample concept map (CF example)
Correct arrows for CF include: gene (CFTR) → undergoes → mutation (F508del) • mutation → produces → altered protein (misfolded CFTR, degraded before reaching membrane) • altered protein → causes → physiological consequence (no Cl− secretion, thick mucus, lung infections) • physiological consequence → is the basis of → genetic disease (cystic fibrosis) • gene → encodes → altered protein. Award 1 mark per correctly labelled, causally directed arrow (max 5).